Conversion of MDL Query Formats
to SMARTS and SMIRKS
Barnard Chemical
Information Ltd., Sheffield UK
This presentation discusses the problems of interconverting not-entirely-compatible
structure query representations. Specifically, the conversion of MDL
Molfiles and RGfiles to SMARTS strings is described,
and issues such as implicit/explicit hydrogens,
ring-embedment,
aromaticity,
link
atoms and R-groups, and the compromises
needed in their conversion, are covered. In addition, the conversion of
MDL RxnFiles to Reaction SMARTS and SMIRKS
strings is described, and algorithms for the automatic
assignment of atom maps are outlined.
MDL's ISIS/Draw
program contains various features for expressing substructure and reaction
search queries:
-
atom types (fully specified, unspecified, list of permitted/forbidden)
-
implicit hydrogen counts
-
number of substitutions permitted
-
atom valence
-
number of ring bonds
-
ignore/require stereo parity match
-
bond types (single/double/triple/aromatic/any)
-
bond stereochemistry
-
ring/chain bond
-
repeated linking groups
-
R-groups
-
etc.
Query structure can be exported as Molfile (simple query)
, RGfile (R-group query) or RxnFile (reaction query).
Descriptions
of these file formats are published on the web, and other vendors have
software products which can write them.
Daylight's
SMARTS language includes similar features (SMARTS atomic and bond primitives)
for similar purposes.
Automatic conversion of the MDL file formats to SMARTS
would allow the convenience and familiarity of MDL's (and other vendors')
drawing programs to be combined with the speed of Daylight's searching.
But subtle differences in the atom and bond properties
which can be expressed in each language cause some problems for interconversion
programs.
MOLSMART program:
Hydrogen Counts and Further Substitution
In MDL files you can specify
-
explicit hydrogens (as separate atoms)
-
minimum number of hydrogens permitted in excess of those
explicitly drawn
-
number of substitutions allowed at an atom
In SMARTS you can specify
-
explicit hydrogens (as separate atoms)
-
total hydrogen count (H)
-
implicit hydrogen count (h)
-
total connections (X)
-
explicit connections (D)
At present the MOLSMART program makes the following conversions:
-
substitution count (M SUB) ->
"D" primitive
-
explicit hydrogen atoms -> explicit hydrogen atoms
-
implicit hydrogen count -> "h" primitive, but to negate possibility
of fewer than specified
e.g. MDL's H2 is converted to ";!h0;!h1"
This is appropriate where explicit hydrogens are used
for "good" reasons:
-
charged hydrogen
-
isotopic hydrogen
-
bridging hydrogens
-
hydrogens connected to another hydrogen
-
hydrogens which are changed during a reaction
Because some people use explicit hydrogens in ISIS/Draw to
prevent further substitution it might be better to convert such explicit
hydrogens in the MDL file to the SMARTS "H" primitive. Comments
on this would be welcome.
Ring and Ring Bond Counts
It is often desirable to specify whether or not a particular
substructure may occur as part of a ring, or a particular ring as part
of a larger ring system.
-
MDL allows you to specify the number of ring bonds attached
to an atom
-
SMARTS allows you to use atomic primitives to specify the
number of SSSR rings an atom occurs in, and the size of the smallest such
ring
-
SMARTS also allows you to use the bond primitive "@"
to specify bonds as being "any ring bond" [N.B. this should not be
confused with the SMARTS atomic primitive for anticlockwise chirality]
If the MDL file specifies "no ring bonds", this can be exactly
converted to the SMARTS "R0".
If the MDL file specifies an exact number of ring bonds,
the MOLSMART program converts this to a recursive SMARTS.
e.g. for a carbon atom specified to have a ring
bond count of 3:
[ C; $ (* (@*) (@*) @* ) ]
This is, however, not totally accurate as it specifies only
a minimum of 3 ring bonds, whereas the MDL file specifies exactly
3. Any suggestions on how
to provide a more faithful translation would be welcome. For 4 ring bonds,
the translation is accurate.
Aromaticity
-
MDL regards aromaticity as a bond property.
Bonds can be specified as "single", "double", "aromatic"
or a combination of these.
-
SMARTS regards aromaticity (largely) as an atom property.
Aliphatic atoms have upper-case symbols,
aromatic atoms have lower-case symbols, and
aromatic bonds are assumed between adjacent aromatic
symbols.
In conversion the program has to deduce whether an atom is
aromatic or not from the bonding pattern.
Aromatic Atoms:
-
Atoms which have at least one aromatic bond.
Aliphatic Atoms:
-
Chain atoms (atoms specified to have no ring bonds)
-
Atoms with more than one implicit hydrogen
-
Atoms with more than one single bond
-
Atoms with either a double bond or a triple bond
-
Atoms with a defined substitution count which is not 2 or
3
-
Atoms with a defined "valence" (total number of bonds) which
is not 3.
-
Atoms which are not B, C, N, O, Al, Si, P, S
Atoms which do not fall into any of the above classes could
be either aromatic or aliphatic. They are shown in SMARTS expressions of
the form "#nn" which effectively encompasses both upper
and lower case forms.
e.g. "#6" for aromatic or aliphatic carbon.
Remaining Problems:
-
Rigid use of the #nn notation for ambiguous
atoms may lead to "inefficient" SMARTS for searching.
How far would we be justified in assuming such atoms
to be aliphatic in the interests of efficient searching?
-
Though it is possible to specify aromatic bonds in MDL files,
it is also possible to show alternating single and double bonds in aromatic
rings. Atoms in such rings will not at present be recognized as aromatic.
We have recently implemented code to convert the bonds in such rings to
"aromatic" automatically.
Should this conversion always be used, or only as a user
option?
Comments are
welcome.
Link Atoms
Indicate nose-to-tail repetition of parts of the structure.
Different conversion strategies are needed if they occur in rings or in
chains.
In chains, we can use recursive SMARTS:

[#7] [ $(C ([#7]) [#7] ),
$(C ([#7]) C[#7] ),
$(C ([#7])CC[#7] ) ]
Note that [#7] is used for the nitrogens,
since either of them might be aromatic or aliphatic. Note also that
[#7] [ $(C[#7]), $(CC[#7]),
$(CCC[#7]) ]
cannot be used as it would match any structure which
just has a NC in it.
In rings, separate SMARTS (to be searched in OR relationship)
are needed, because we cannot have matching ring closure symbols inside
and outside the recursive SMARTS:

N1 C N1
N1 CC N1
N1 CCC N1
Here the nitrogens have two single bonds, and so must
be aliphatic.
R-Groups
ISIS/Draw permits sophisticated R-group queries to be constructed,
which can be searched against databases with the ISIS
Power Searching Module. Complex "logic" can be used to define the relative
frequency of occurrence of different R-groups at different positions.
In simple cases, a single recursive SMARTS can be produced:
|
R1-C-C-R2
|
R1 = Cl or NO2
R2 = ethyl |
C ( [ $(N(CC)(=O)=O), Cl ] ) C [C;D2] [C;D1]
In more complex cases involving multiple occurrence of
R-groups, and involved "logic", it may be necessary to produce a large
number of separate SMARTS (effectively, one for each possible combination
of R-group members). e.g. no less than 80 different SMARTS are needed for
this query:

Reaction SMARTS
ISIS/Draw can be used to specify reaction queries; these
can be exported to RxnFiles.
With the use of the reaction arrow (>>)
and appropriate top-level parentheses these can be converted to reaction
SMARTS queries.
There is some simplification because some of the MDL features
(such as R-groups) are not permitted in reaction queries.
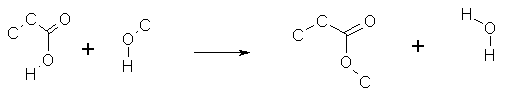
([#6]CC(=O)O[H]).(O([#6])[H])
>> ([#6]CC(=O)O[#6]).(O([H])[H])
Atom map classes can be specified in ISIS/Draw.
If they are provided, they are copied to the reaction
SMARTS, though for internal reasons the actual numbers used are usually
changed.
SMIRKS
-
SMIRKS
is a restricted version (subset) of reaction SMARTS.
-
It represents generic reactions ("transforms") and can be
used to generate specific reactions and products from precursor molecules
-
It can be laborious to generate manually
-
MOLSMART can check whether or not the SMARTS it is generating
conforms to SMIRKS requirements.
-
This was complicated by Daylight changing
the rules for SMIRKS.
Automatic atom mapping
One SMIRKS requirement is that atoms appearing on both sides
must be mapped (unmapped atoms are assumed to disappear or appear).
To avoid the need for the user to map the reaction manually
in ISIS/Draw, MOLSMART includes an algorithm to assign these maps automatically.
This is based on the Principle of Minimum Chemical Distance
(MCD):
-
Algorithm tries different ways of mapping reactant atoms
to product atoms
-
Chooses the one which involves fewest bond changes during
reaction
-
No need to complete the proposed mapping if it can be seen
that it will result in a larger chemical distance than the best found so
far.
-
Various heuristics used to help find "good" mappings early
on.
-
Most time is required to "prove" that there really aren't
any better ones - to save time this is terminated after a certain number
of iterations
-
"Less than optimal" mappings are still valid, but will involve
more bond breaks/formations than are necessary
The current version requires a fully balanced reaction for
mapping.
-
not a problem when SMIRKS had to be fully balanced
-
but then Daylight went and changed the rules
-
program adds "trivial" reagents and by-products
to balance reaction if needed
-
these are not added to the final SMIRKS
but used only to help mapping algorithm
We have been working to extend the mapping algorithm to handle
unbalanced reactions.
-
initially tried finding the "maximal set of most common substructures"
(equivalent to finding the MCD for balanced reactions)
-
now trying a simpler idea based more directly on MCD
-
prototype implementation looks promising
-
strange results can occur where molecules with common substructures
are missing on both sides of the reaction.
Applications using MOLSMART are being discussed at MUG99
by Pat
Walters and Meixiao
Liu.
A scaled-down version of MOLSMART (for SMIRKS generation
only) has been incorporated into the latest release of MSI's
WebLab Diversity Explorer.
Acknowledgements:
-
Annette von Scholley-Pfab

-
Abbott Labs (Meixiau Liu, Jerry Delazzer, Randy Chen)
-
Daylight (Jeremy Yang, Jack Delaney)