



Analysis of Results(progress report)
-
Comparisons of predicted vs observed binding geometries.
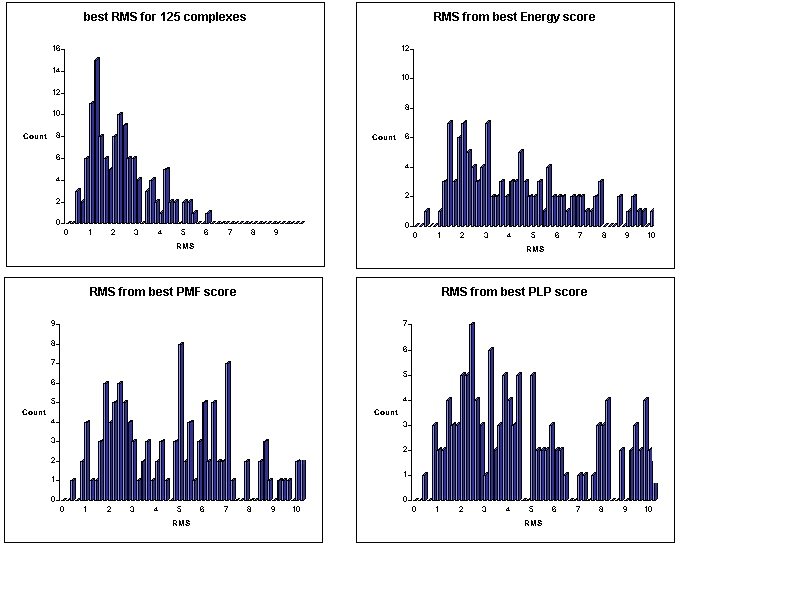
-
Generally, the DG method generates structures close to the crystal structure. Mean for the best RMS is 2.1 angstroms.
-
Individual scoring methods don't reliably pick out the best structures.
-
The three methods seldom all make large errors on the same protein.